
A Simulation of Tungsten Hexacarbonyl Matrix Isolation
Abstract
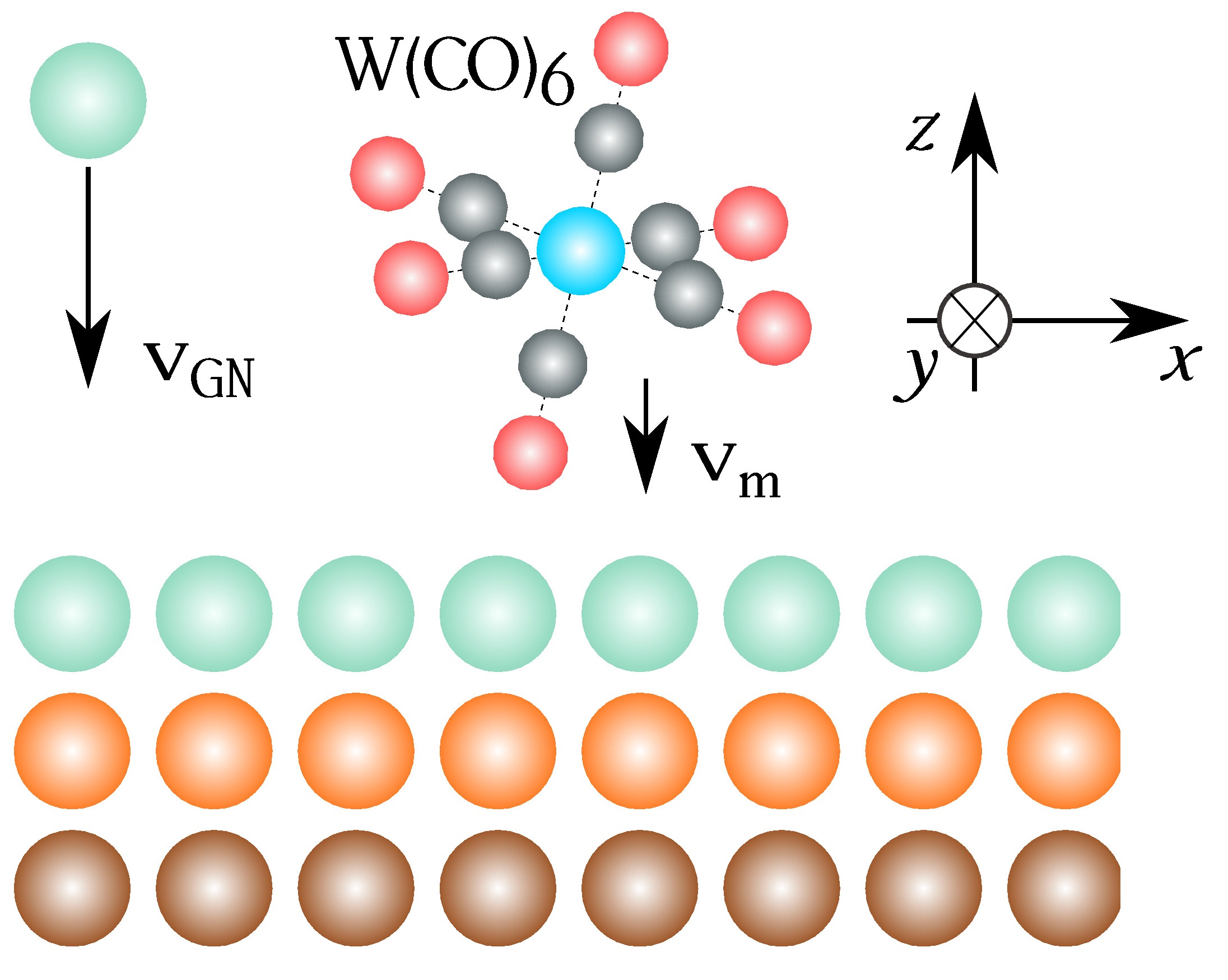
We use a deposition model in order to simulate the grow of rare gases crystals doped with tungsten hexacarbonyl as impurity. The simulations were carried out using Classical Molecular Dynamics, applying quantum corrections to improve the interactions between atoms. Several trapping sites were identified and probabilistic and energetic studies were done as well. We found that sites preference of the molecule is close related with crystallographic properties of rare gases solids and the temperature of the system. The obtained information is quite important to clarify the experimental results obtained by trapping the molecule using matrix isolation technique and infrared spectroscopy. Results were compared with experimental data recorded from previous studies of the system.

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.
This work is licensed under the Creative Commons Attribution-NonCommercial 4.0 International (CC BY-NC 4.0) license.







